| 双或多靶点 CRISPR/CAS9 敲除系统使用教程 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 发布时间:2018-06-28 23:59:00 | 浏览次数: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
以下只是介绍双靶点的敲除系统的构建方法,如需要多靶点敲除,选择酶切位点组合添加多个U6-sgRNA即可。
Gene Knockout Mediated by CRISPR-Cas9 System (Cas9-2hitKO)
CRISPR-Cas9系统简介:
图1. CRISPR-Cas9 系统介绍
CRISPR-Cas9 系统是一种被广泛运用基因组编辑工具,它来源于细菌的适应性免疫系统。CRISPR-Cas9系统包括:Cas9酶 和一个向导 RNA。 向导RNA 作用是引导 cas9 到基因组的特异性位点上切割。如图1所示。目前为止,CRISPR-Cas9 系统主要有两方面应用: 基因敲除和基因敲入。基因敲除时, 一旦DNA的双链断裂反应(DSB)被Cas9切割诱导发生, 细胞会启动 NHEJ DNA修复方式,这会造成DNA的删除和插入。对于基因敲入,加入一个可同源重组的DNA片段, 细胞会将这个一段DNA片段插入进基因组 。
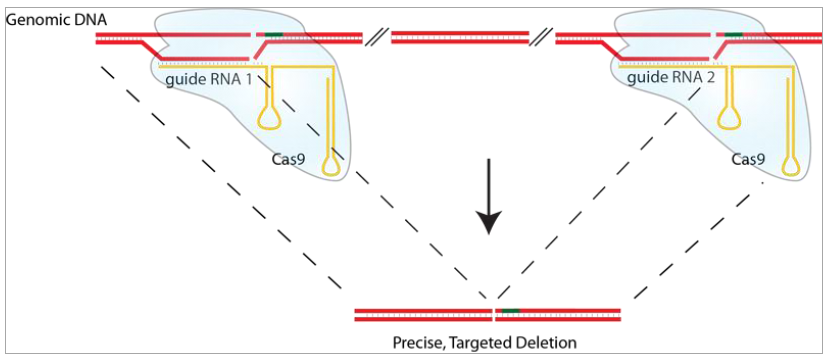
两次或者三次切割介导的基因敲除
与先前的敲除策略并不一样,我们改进的这个操作系统可以将基因的删除做到可控的特定长度。我们正在基因组的一个特定的区域引入两个或者三个sgRNA。这些sgRNA指引Cas9酶在这两个位点进行切割。然后切割后末端通过 NHEJ连接上。通过这个策略,我们可以将一个基因的独立外显子或者整个基因敲除掉。如图2所示。
图2. Cas9-2hitKO系统 
图3. Cas9-3hitKO系统 Cas9-2hitKO系统中涉及到载体 为了达到高效的敲除效率,我们在同一个载体中引入cas9 酶和两个向导性RNA。整个系统包括3个载体:pX458M ,pX459M和2个过度载体(pHH-hU6-A-gRNA,pHH-hU6-B-gRNA)。pX458MCS ,pX459MCS除了含有Cas9和一个向导性RNA插入位点,还引入一个36碱基的多克隆位点。过度载体是为插入第二个或者第三个sgRNA 的辅助性载体。
图4. PX458M图谱
图5. PX459M图谱
PX458M和PX459M 载体是从张峰实验室 PX458 and PX459 载体改造过来的。主要是引入36个碱基的多克隆位点(详细序列见pX458M和pX459M网页)。PX458M 带有 EGFP 荧光标记; PX459M带有 puromycin 抗性筛选标记。
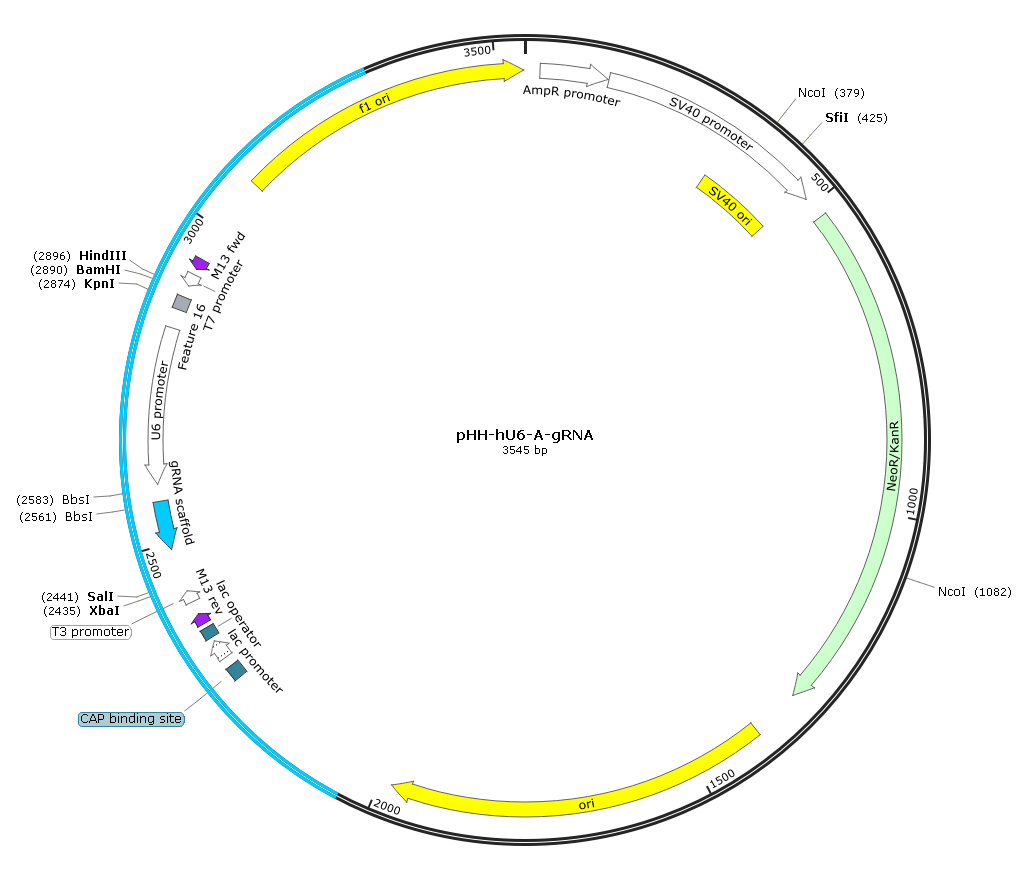
图6.中间质粒pHH-hU6-A-gRNA图谱
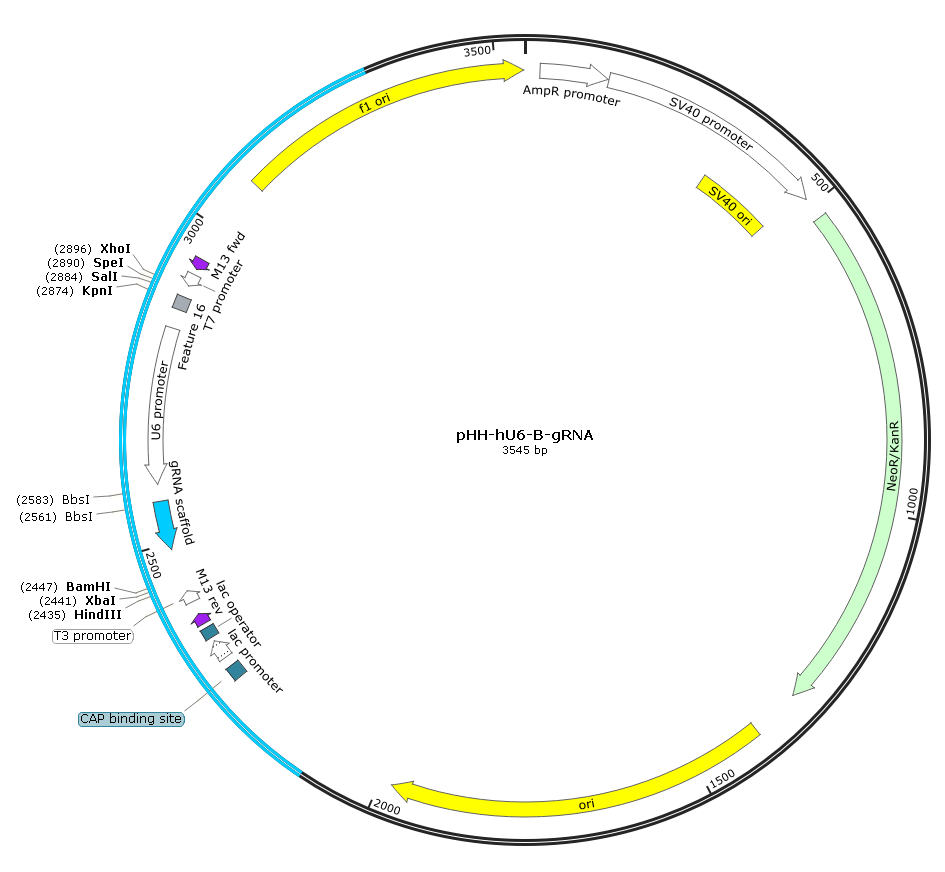
图7.中间质粒pHH-hU6-B-gRNA图谱
过渡载体由addgene #53188 phU6-gRNA,在“hU6-gRNA scaffold”两侧添加酶切位点改造而来,用酶切整个“hU6-sgRNA-gRNA scaffold”,可用于多靶点CRISPR 敲除系统,也可以单独用于表达sgRNA。
Guide RNA插入
根据张峰实验的实验操作方法, guide RNA 插入方法很简单。如图6所示,可以用 BbsI消化PX458M或者PX459M载体, Guide RNA是通过引物退火方法(20 bp 长)引入的BbsI粘性末端通过T4 DNA连接酶插入到载体中。
图8.向导性 RNA插入方法
实验流程
I. 设计脱靶效应低Guide RNA序列
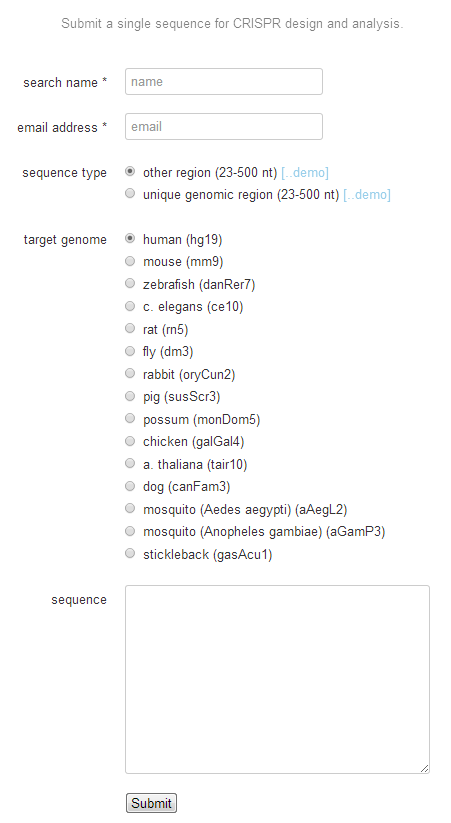
基于网站的GuideRNA的设计 网站地址:http://crispr.mit.edu/. 基本的流程如下图。
1. 输入最多250个核苷酸的基因组 DNA序列进 图7的Sequence框中 选择相应的物种。点击‘submit’。 2. 点击‘Guides & offtargets’. 选择得分90分以上的序列。序列的方向并不重要,两个方向都可以选择。
图9. Guide RNA 设计流程
II. 选择一对GuideRNA来敲除基因 敲除基因组一段序列,选择一对 Guide RNA就显得非常重要。根据前人的实验,敲除基因 1M b并没什么问题。我们一般建议敲除10Kb以下。 选择一对Guide RNA的标准如下, a. 如果目的基因比较短,在10kb 左右 ,建议敲除全基因,甚至包括启动子。 b. 如果目的基因比较长,建议敲除比较重要的外显子。 c. 目的基因比较长,也可以敲除全部的启动子和部分的基因编码区 d. 如果目的基因在发表的文献中已经报导过用传统方法敲除,我们可以用这套系统敲除同样区域。
III. 构建双Guide RNA克隆
1. 合成引物.
前向引物: CACC-(N)20 方向引物: AAAC-(N)20R 如果根据张峰网站设计的Guide RNA第一个核苷酸不是‘G’,在5’端补加一个 ‘G’,因此设计好引物变成:
前向引物: CACC-G(N)20 反向引物: AAAC-(N)20RC
2. 引物磷酸化与退火
95℃ 5min,自然降温到室温(或者用PCR梯度降温)。
将载体酶切尽可能保证载体全部双酶切切开,可以选择NEB没有星活性的的酶。
取1ul 退火后的溶液,与载体(20-50ng就够了)连接并转化。
3. Px458M(或 PX459M) 和pHH-hU6-gRNA载体的 BbsI酶消化
割胶回收载体,回收后的载体浓度应大于50 ng/ul。
4. 连接反应
16 ℃连接过夜
5. 转化和鉴定阳性克隆 转化后挑取单克隆LB培养,然后PCR鉴定克隆。Px458M 或Px459M载体 用hU6测序。而pHH-hU6-gRNA, 用 M13F /R作为鉴定引物测序。
6. 将Guide RNA2 从pHH-hU6-gRNA转入到Px458M /Px459M-Guide RNA1得到插入双Guide RNA的载体Px458M /Px459M-Guide RNA1-Guide RNA2(以pHH-hU6-A-gRNA为例,也可以选用A的其他酶切位点,或者不同酶切位点的pHH-hU6-B-gRNA)
7. 割胶回收
8. 连接反应
16 ℃过夜连接。
IV. 如果是敲人源细胞中的基因,我们可以在293FT细胞检测 系统的敲除效率,小鼠的细胞中的基因可以在MEF和NIH3T3中检测系统的敲除效率 1. 将上述Px458M /Px459M-Guide RNA1-Guide RNA2转染进293FT细胞中 36-48 小时 2. 通过荧光显微镜和流式细胞仪检测转染效率 3.提取转染细胞基因组,进行基因型鉴定。引物的设计方案如图11所示。图12显示敲除 PPM1A 基因。
V. 敲除自己感兴趣的细胞中的基因 理论中,只要转染进Px458M /Px459M-Guide RNA1-Guide RNA2载体的细胞就是基因敲出的细胞,然而我们想得到纯的基因敲除细胞株需要进行细胞单克隆化培养。对于一些细胞,单克隆化培养并不是那么容易。为了增加单个细胞的扩增,我们可以预先包被gelatin和条件性诱导培养液。
1. 细胞提前一天铺在6孔板中,确保细胞密度在70%-80%。 2. Px459M-Guide RNA1-Guide RNA2转染自己感兴趣的细胞并培养24 小时。 3. 用puromycin连续筛选3到4 天。 以没有转染的细胞做对照。 注意!: 每种细胞筛选的puromycin并不完全一样,应该通过梯度实验摸索最佳浓度 4. 撤销 puromycin, 用 20% FBS培养液培养细胞。 5. 几天后,如果有克隆发生增殖了,挑取克隆,并转移到24孔板中继续培养。剩下的细胞也可继续培养。 6. 24孔板中细胞达到 30%密度, 胰酶消化细胞,取出 80% 细胞来做基因型鉴定, 20% 细胞继续培养。 7. 14000rpm离心收取细胞30s。加入 20-60ul solution A裂解细胞。95 ℃加热10 min,然后加入同样体积的 solution B, 涡旋震荡一下, 13000rpm 离心0min。取上清做基因型鉴定的模板。 8. 做完基因型鉴定后,只继续培养基因敲除的细胞克隆。 9. 当基因敲除的细胞克隆长满时,胰酶消化,进行细胞计数。以40个细胞每10毫升进行10倍梯度稀释。 10. 第9 步中培养液也就是条件刺激培养液,也就是收集第9步中培养时间少于24小时的旧的培养基,在这样的培养基中加入20% FBS 后用 0.22 ul 滤膜过滤后的细胞培养液称为条件刺激培养液。 11. 在96孔中,每孔加入100 ul 除菌的0.1% gelatin溶液, 在37 ℃ 细胞培养箱中放置30分钟以上。 12. 吸去 gelatin,条件刺激的细胞培养液重悬细胞,按每孔100 ul,大约 0.4 cell 加入到96孔每个孔中。加入8 到10 个孔。摇匀继续培养。 13. 4到5天后在上述孔中继续加入条件刺激培养液100 ul每孔,不要超过5天。 14. 当细胞密度超过 30%, 或者克隆长得足够大,将它转移到24孔板中继续培养。 重复6-8步。 15. 步骤5中剩余的细胞长到50% 密度时,继续有限稀释进行单克隆化培养。也可以冻存起来留置后用。 VI. 评估脱靶效应 脱靶的情况在网址 (http://crispr.mit.edu/.) 上评估。这个网站上列举上很多潜在的脱靶位点。把这些位点通过PCR的方法扩增出来进行PCR产物测序。如果没有发现建立好基因敲除细胞株有明显的脱靶情况,那么这样的细胞可以用来进行后续的实验。
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 上一篇:VIGS-操作基本方法 下一篇:荧光蛋白特性 |