| shRNA详细教程-以pLKO.1-Puro为载体设计 |
| 发布时间:2018-05-03 09:29:54 | 浏览次数: |
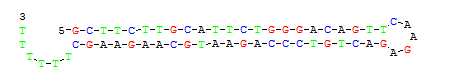 |
shRNA设计此教程用于不知道siRNA靶点的设计,如果已经从文献中获得序列,可以从第5步开始设计
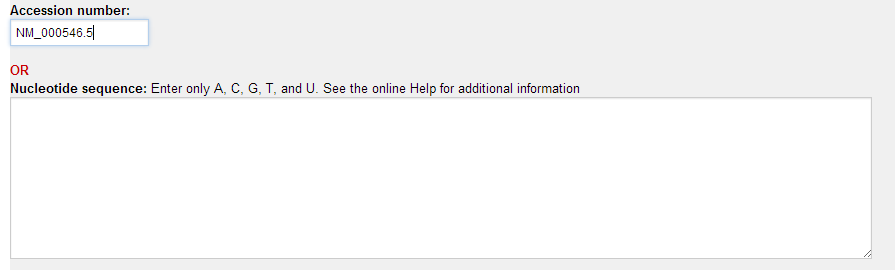
1.选择基因 例如 Gene Name: TP53 Accession: NM_000546.5
2. 寻找靶点 输入基因序列号,或者粘贴序列
点击 RNAi设计
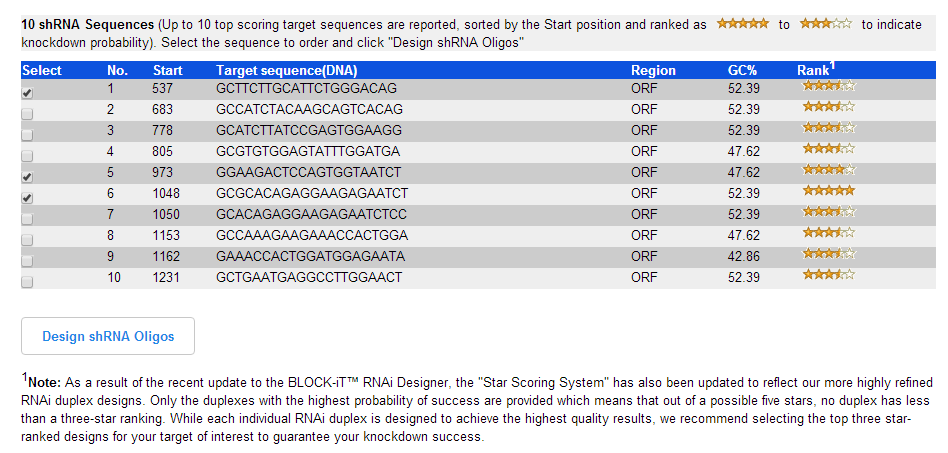
3.靶点选择 (选择星号比较多的2-3个作为目的序列)
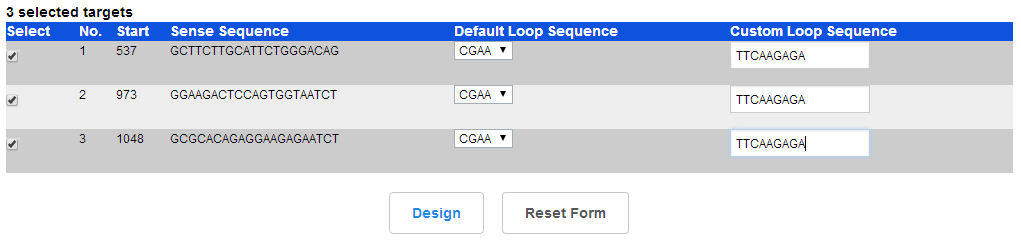
4.shRNA网页设计 点击设计shRNA OLigos
可以在 网页上的loop 也可以不更改(后面需要更改为常用loop序列TTCAAGAGA) 点击设计
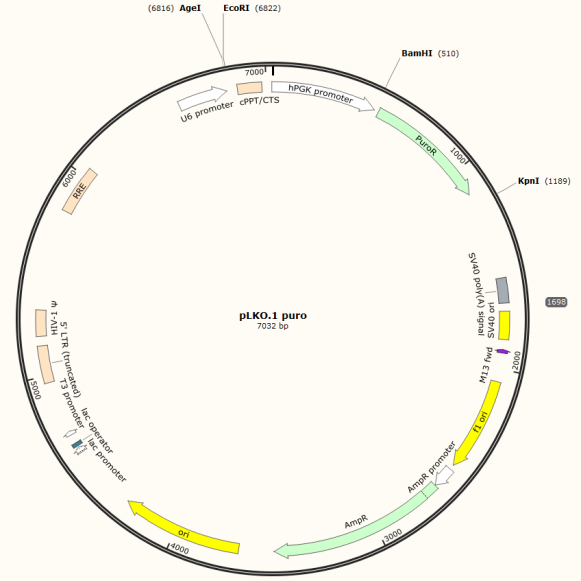
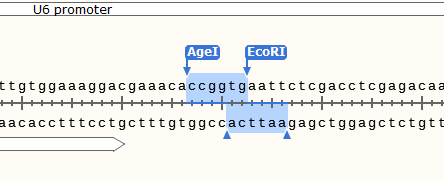
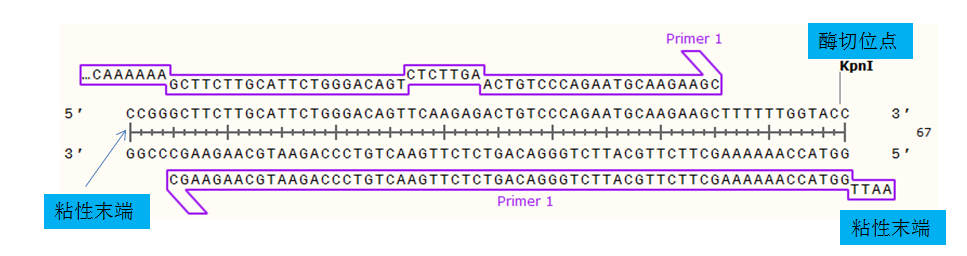
5.后续的根据自己的载体手工设计shRNA (以PLKO.1-puro设计,酶切位点AgeI EcoRI,酶切位点后1300bp有一个单一的酶切位点KpnI)
注意:shRNA比较短,而且结构复杂,测序困难,选择载体上的单一酶切位点,设计到shRNA后面,用于后续的单酶切验证。
设计模板按照下图 (下图去除了AgeI和EcoRI酶切位点,也可以适当位置添两个碱基保留着两个酶切位点,以后还可以当做载体用)
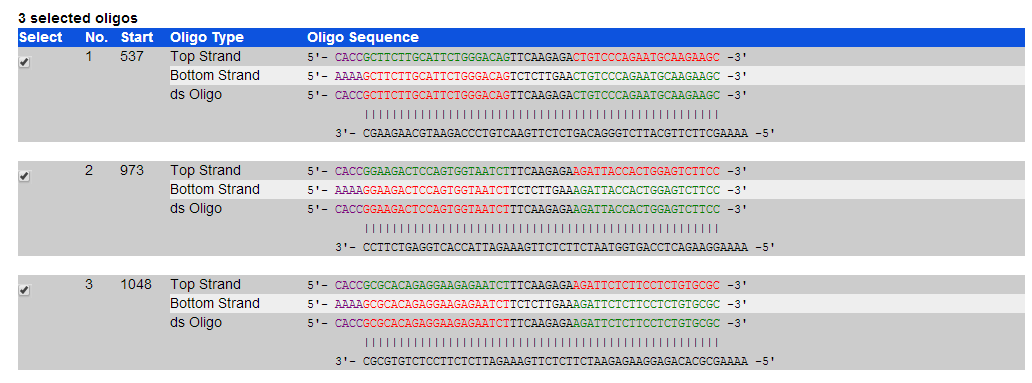
5’-CCGG siRNA正向序列TTCAAGAGA 反向互补序列TTTTTT GGTACC-3’ 3’- 正向互补序列 AAGTTCTCT 反向序列 AAAAAA CCATGG TTAA-5’
将网页上设计的shRNA 序列替换到上述模板 直接将ds Oligo的非突出的序列复制替换到上述的模板上
替换之后如下
5’-CCGG GCTTCTTGCATTCTGGGACAGTTCAAGAGACTGTCCCAGAATGCAAGAAGCTTTTTT GGTACC-3’ 3’- CGAAGAACGTAAGACCCTGTCAAGTTCTCTGACAGGGTCTTACGTTCTTCGAAAAAA CCATGG TTAA-5’
注意:正义链为5’→3’,反义链为3’ →5’,合成时需要进行反向处理。
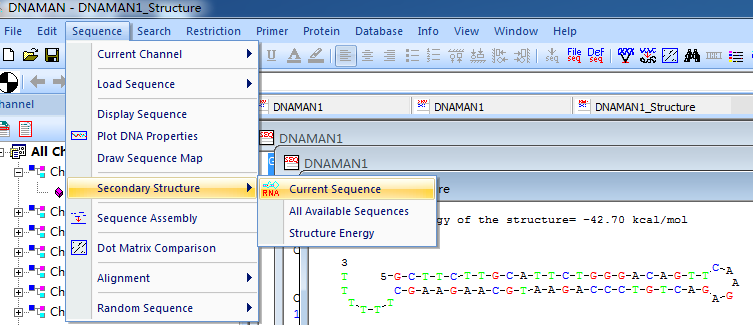
6.验证设计的shRNA是否合适 二级结构(用DNAMAN分析二级结构)
分析正反向引物是否匹配 用Snapgene 分析,正向序列输入进去,反向的作为引物用来验证(注意5’ 3’方向) 一个正确的shRNA应该入下图
7.最终发送合成的序列为 5’-CCGG GCTTCTTGCATTCTGGGACAGTTCAAGAGACTGTCCCAGAATGCAAGAAGCTTTTTT GGTACC-3’ 5’- AATTGGTACCAAAAAAGCTTCTTGCATTCTGGGACAGTCTCTTGAACTGTCCCAGAATGCAAGAAGC -3’
8.引物合成 shRNA对引物纯度要求不高,不需要用page纯化,为了节约成本,用低一级的纯化方式HAP即可使用。
9.引物退火连接 稀释到50uM 取各取4.5ul,加1ul,退火buffer(可以用不含dNTP和酶的PCR buffer),95℃ 3min,自然降温到室温。
将载体酶切尽可能保证载体全部双酶切切开,可以选择NEB没有星活性的的酶。
取1ul 退火后的溶液,与载体(20-50ng就够了)连接并转化。
10.质粒提取并验证 如果有载体自连对照为阴性,可以取两个单克隆提质粒KpnI单酶切,如果有能切出一个1300bp左右的片段,可以认为构建是正确的。 取一个质粒进行测序,测序的时候告知测序公司,此质粒为发卡结构,和poly结构。
附录1. shRNA启动子 多数的siRNA表达载体依赖RNA聚合酶III 启动子(pol III)中的一种,操纵一段45—50nt的发夹结构RNA(small hairpin RNA,shRNA)在哺乳动物细胞中的表达,shRNA在细胞内会自动被加工成为siRNA,从而引发基因沉默或者表达抑制.这一类启动子包括大家熟悉的人源和鼠源的U6启动子和人H1启动子等。采用RNA pol III启动子的原因是由于它有明确的启始和终止序列,总是在离启动子一个固定距离的位置开始转录合成RNA,遇到4—5个连续的U即终止,并且转录产物在第二个尿嘧啶处被切下来,非常精确,而且还可以在哺乳动物细胞中表达更多的小分子RNA.转录出的RNA形成发卡样结构后,会在3’端形成2个突出的尿嘧啶,这类似于天然的siRNA,因而有利于双链RNA诱发RNAi。要使用这类载体,需要订购2段编码短发夹RNA序列的DNA单链,退火后克隆到相应载体的pol III 启动子下游。克隆的过程需要几天的时间,也需要经过测序以保证克隆的序列是正确的。 先在体外构建能表达双链RNA的载体,再将载体转移到细胞内在细胞内合成出双链RNA,不但能增加有效转染细胞的种类,而且在长期稳定表达载体的细胞株中,双链RNA能够长期发挥阻断基因的作用。 现在有多个报道说通过质粒表达siRNAs同样可以抑制特定基因的表达.当这种带有PolIII 启动子和shRNA编码序列的质粒转染哺乳动物细胞时,这种能表达siRNA的质粒确实能够下调特定基因的表达,抑制范围包括外源基因和内源基因。
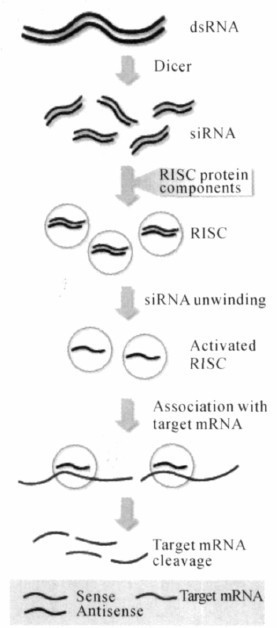
附录2 RNAi的作用机制及siRNA的合成方法 RNA干扰(RNA interference,RNAi)是由双链RNA(double strandedRNA, dsRNA)分子在mRNA水平关闭相应序列基因表达或使其沉默的过程。dsRNA可以抑制不同类型细胞的靶向基因表达,用特异性的抗体几乎检测不到靶向基因所表达的蛋白质。因此,RNAi技术又被形象地称为基因敲除(knock out)或基因沉默(gene silencing)。RNAi是一种典型的转录后基因调控方法,又称转录后基因沉默(post tracriptional gene silencing,PTGS)。 1 RNAi技术的发展过程 早在1990年,植物学家Jorgeen等人用矮牵牛花做试验,将紫色素合成基因导入植物体,尝试将更多拷贝的色素基因注入植物体,使花朵的色彩更加艳丽。结果许多花朵没有开出更艳丽的花朵,反而开出白色的花朵。进一步分析发现,这些转入的基因不但自身没有表达为蛋白质,反而关闭了牵牛花中与其同源的色素相关基因的表达。由于这种由外源基因的导入而造成的抑制作用最初被确认是发生在转录后水平,称这种现象为共抑制(cosureion)。1994年,Cogoni等人将外源性类胡萝卜素基因导入野生型粗糙链孢霉菌,结果转化细胞中内源性的类胡萝卜素基因也受到了抑制,他们称这种基因失活形式为消除作用或基因压制(quelling)。1995年康乃尔大学的Guo博士在实验中想通过反义RNA阻断美丽线虫(C. elega)的par-1基因表达,同时还用正义RNA做了一个对照,试图观察到对照试验组基因表达增强的现象,但结果却发现了反义和正义RNA都阻断了该基因的表达,Guo等人一直不能解释该现象。直到1998年Fire等才发现这是由于Guo博士在试验中污染了双链RNA而引起的。他们还证明转录得到的单链RNA经纯化后注射线虫所引起的阻断作用十分微弱,而经纯化的双链RNA则能高效特异地阻断相应基因的表达,他们将此现象称为RNA干扰。1999年, Hunter等的实验进一步验证了RNAi的存在。他们将dsRNA去除后,再将正义或反义的RNA注入线虫卵中,没有产生基因沉默现象。相反,如果将上述正义及反义RNA混合,经退火复性后得到的dsRNA注入线虫卵中可以显著地产生基因沉默现象,从而印证了Fire等提出的RNAi作用是由dsRNA引起的结论。2000年首次在果蝇中发现,通过核糖核酸酶可将长的dsRNA处理为21~23 nt的短片段。2001年,首次报道了哺乳动物细胞中也有RNAi。2002年证明了用短的发夹结构RNA(shRNA)在哺乳动物细胞中可以诱导特异性的基因沉默。2003年首次用RNAi技术对模型动物进行了治疗研究。2006年诺贝尔医学奖没有坚持研究成果要经过数十年实践验证的“惯例”,破格授予美国科学家安德鲁•菲尔和克雷格•梅洛,以表彰他们1998年发现了RNAi现象。此后,人们在不同种属的生物中进行了广泛而深入的研究,结果证实dsRNA介导的RNAi现象存在于真菌、果蝇、拟南芥、锥虫、涡虫、水螅、斑马鱼、小鼠、大鼠、猴乃至人类等多种生物中。 2 RNAi的作用机制 目前关于基因沉默的假说认为,转录后水平的基因沉默,主要包括起始阶段、效应阶段和倍增阶段。 2.1 起始阶段 外源性导入或由转基因、转座子、病毒感染等多种方式引入双链核糖核酸(dsRNA), 在细胞内特异性与RNA酶Ⅲ(RNAaseⅢ核酸内切酶) Dicer结合,dsRNA被切割成21~23nt长度的带有3′端单链尾巴及磷酸化的5′端的短链dsRNA,即小干扰RNA(siRNA)。(以下图片均来自于陈莉等的文章《在医药领域中RNA干扰研究进展》)
2.2 效应阶段 双链siRNA可以与含Argonauto(Ago)蛋白的核酶复合物结合形成RNA诱导沉默复合体(RNA-induced silencing complex,RISC)并被激活。在ATP供能情况下,激活的RISC将siRNA的双链分开,RISC中核心组分核酸内切酶Ago负责催化siRNA其中一条链去寻找互补的mRNA链,然后对其进行切割。反义链先与同源mRNA配对结合,然后RISC在距离siRNA 3'端12个碱基的位置将mRNA切断降解,从而阻止靶基因表达,使基因沉默。
2.3 倍增阶段 siRNA在RNA依赖性RNA聚合酶(RdRP)的作用下,以mRNA为模板,siRNA为引物,扩增产生足够数量的dsRNA作为底物提供给Dicer酶,产生更多的siRNA, 可再次形成RISC,并继续降解mRNA,从而产生级联放大效应。并作用于靶mRNA。如此反复倍增,从而使RNAi的作用进一步放大。因此少量的siRNA就可以产生高效的基因沉默效果。 3 RNAi的设计及合成 3.1 siRNA的设计 设计高效率的siRNA首先要通过NCBI、DDBJ、BMBL这3个基因序列数据库,检索相关的基因序列,获得靶向mRNA或cDNA序列,再就是合理设计siRNA。常规siRNA的设计原则是以成熟的mRNA为标准,若以DNA序列为参照,则最好选择cDNA转录区+50到+100以后的下游序列,两端的非翻译区不作为siRNA设计依据。siRNA二聚体的正义链和反义链各含21nt为佳,3'端为突出末端。3'凸端为尿苷(U),5'凹端为腺苷(A)的mRNA序列应作为首选。进行Gen-Bank BLAST查询,确保所选siRNA编码不与其它基因同源。不是mRNA的所有亚单位都能形成有效的siRNA,大约有60%~70%可以发挥沉默效应, 这是由于5‘和3’-UTR有丰富的调控蛋白结合区域,产生UTR结合蛋白或翻译起始复合物均可能影响RISC结合mRNA,从而影响siRNA的效果,所以沉默效应率差异较大,针对一个靶基因需要设计3~4条siRNA,以保证能够筛选出一条有效序列。 3.2 siRNA的合成方法 制备siRNA的方法主要有化学合成法、体外转录法、酶消化法、体内转录法等。当已经找到最有效的siRNA时,适合以核苷酸单体为原料,通过化学方法合成两条互补的长约21~23 nt的RNA单链,然后退火形成双链复合体,这种方法所获得的产物纯度、干扰效率高,合成简单,但是制备周期长,作用时间短,而且价格昂贵限制了其应用和推广。体外转录法是以两段互补的DNA为模板, 在针对靶序列的正义链和反义链上游接上T7启动子,使用T7 RNA聚合酶,各自在体外转录获得两条单链RNA,将两条单链退火后形成双链RNA,然后用RNA酶消化,所得产物经纯化后就是所需的双链RNA,此方法适用于筛选有效的siRNA,但不适用于对特定siRNA进行长期研究。酶消化法是先在体外化学合成200~1000 完整的目的基因长片段dsRNA, 用细胞内形成的siRNA关键酶-Dicer酶或者RNaseⅢ进行消化,即可得到各种不同的针对同一目的基因的不同位点的siRNA混合物,然后将混合物转染至细胞内,能得到较高的抑制效率。该方法抑制率高,且省时省力, 但此法产生的混合物可能导致非特异性抑制,另外在体外转录长链RNA有困难,Dicer酶费用较高,此方法不适用于长时间的研究项目,或者是需要一个特定的siRNA进行研究,特别是基因治疗。体内转录是将siRNA的质粒、病毒表达载体或带有siRNA表达盒的PCR产物转入细胞,由细胞表达产生RNA干扰作用。siRNA表达载体常用能在哺乳动物细胞中表达shRNA的含有RNA聚合酶Ⅲ(polⅢ)启动子的载体,通过转染含有RNA聚合酶II/III的启动子U6或H1,及其下游一小段特殊结构的质粒或病毒载体到宿主细胞内,转录出shRNA,该RNA在细胞内被Dicer酶剪切成siRNA而发挥作用。该方法的优点是细胞特异性强,适用于基因功能的长时间研究。siRNA表达框(siRNA expreion caettes, SECs)是一种由PCR制备的siRNA表达模板,可直接转入细胞进行表达而无需事先克隆到载体中。利用引物延伸法进行PCR,产生包含1个RNA聚合酶III启动子U6或H1、一小段编码shRNA的DNA模板和1个RNA聚合酶III终止位点的表达框架,然后直接转染到细胞内。该方法为筛选有效的siRNA片段和合适的启动子提供了较为便捷的工具。其主要缺点是PCR产物转染细胞的难度大,如有转染试剂能提高SECs的转染效率,这一方法将得到更为广泛的应用。
|
| 上一篇:CRISPR 下一篇:资料资源下载 公众号关键词回复对照表(更新中) |